The
Journal of Chemical Physics 107, 902-913 (1997)
Tino G. A. Heijmena, Tatiana Koronab, Robert Moszynskib,
Paul E. S. Wormera, and Ad van der Avoirda
a Institute of Theoretical Chemistry, NSR Center, University
of Nijmegen, Toernooiveld 1, 6525 ED Nijmegen, The Netherlands
b Department of Chemistry, University of Warsaw, Pasteura
1, 02-093 Warsaw, Poland
Ab initio potential-energy surface and rotationally
inelastic integral cross sections of the Ar-CH4 complex
Abstract
Symmetry-adapted perturbation theory has been applied to compute the
intermolecular potential-energy surface of the Ar-CH4 complex.
The interaction energy, including high-level intramonomer correlation effects,
is found to be dominated by the first-order exchange contribution and the
dispersion energy. The ab initio potential has four equivalent minima of
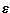 m=-144.30 cm-1 at Rm=7.00 bohr, for structures
in which the argon atom approaches the face of the CH4 tetrahedron.
The computed potential-energy surface has been analytically fitted and
used in converged close-coupling calculations to generate state-to-state
integral cross sections for rotational excitation of CH4 in collisions
with argon. The computed cross sections are generally in good agreement
with the experimental data [W. B. Chapman et al., J. Chem. Phys. 105, 3497
(1996)]. Some discrepancies for the smallest cross sections can be explained
by the influence of sequential collision channels, with the use of a master
equation approach.
m=-144.30 cm-1 at Rm=7.00 bohr, for structures
in which the argon atom approaches the face of the CH4 tetrahedron.
The computed potential-energy surface has been analytically fitted and
used in converged close-coupling calculations to generate state-to-state
integral cross sections for rotational excitation of CH4 in collisions
with argon. The computed cross sections are generally in good agreement
with the experimental data [W. B. Chapman et al., J. Chem. Phys. 105, 3497
(1996)]. Some discrepancies for the smallest cross sections can be explained
by the influence of sequential collision channels, with the use of a master
equation approach.
Back to the publication list of
Tatiana Korona
Tatiana Korona 2003-01-17