Journal of Physical Chemistry A 101, 4690-4698
(1997)
Robert Moszynskia,b, Tatiana Koronab, Paul
E.S. Wormera, and Ad van der Avoirda
a Institute of Theoretical Chemistry, NSR Center, University
of Nijmegen Toernooiveld 1, 6525 ED Nijmegen, The Netherlands
b Department of Chemistry, University of Warsaw, Pasteura
1, 02-093 Warsaw, Poland
Ab Initio Potential Energy Surface and Infrared Spectrum
of the Ne-CO Complex
Abstract
Symmetry-adapted perturbation theory has been applied to compute the
intermolecular potential energy surface of the Ne-CO complex. The interaction
energy is found to be dominated by the first-order exchange contribution
and the dispersion energy. The ab initio potential has a single
minimum of 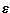 m=-53.49 cm-1 at Rm=6.34 bohr
and \thetam=92.2°. The computed potential energy surface
has been analytically fitted and used in converged variational calculations
to generate bound rovibrational states of the 20Ne-CO complex
and the infrared spectrum corresponding to the simultaneous excitation
of vibration and internal rotation in the CO subunit within the complex.
The computed frequencies of the infrared transitions corresponding to the
m=-53.49 cm-1 at Rm=6.34 bohr
and \thetam=92.2°. The computed potential energy surface
has been analytically fitted and used in converged variational calculations
to generate bound rovibrational states of the 20Ne-CO complex
and the infrared spectrum corresponding to the simultaneous excitation
of vibration and internal rotation in the CO subunit within the complex.
The computed frequencies of the infrared transitions corresponding to the
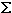
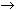 ,
,
 , and
,
subbands are in good agreement with the experimental data [R. W. Randall
et al., Mol. Phys. 79, 1113 (1993)]. The observed bending combination
band is assigned to the transitions from the ground state to the first
excited state. Frequencies of the
, and
,
subbands are in good agreement with the experimental data [R. W. Randall
et al., Mol. Phys. 79, 1113 (1993)]. The observed bending combination
band is assigned to the transitions from the ground state to the first
excited state. Frequencies of the
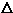 and
transitions which were observed in the static cell spectrum are also reported.
and
transitions which were observed in the static cell spectrum are also reported.
Reprints are available on request. Send an e-mail to
tatiana.korona@tiger.chem.uw.edu.pl
Back to the publication list of
Tatiana Korona
Tatiana Korona 2003-01-17